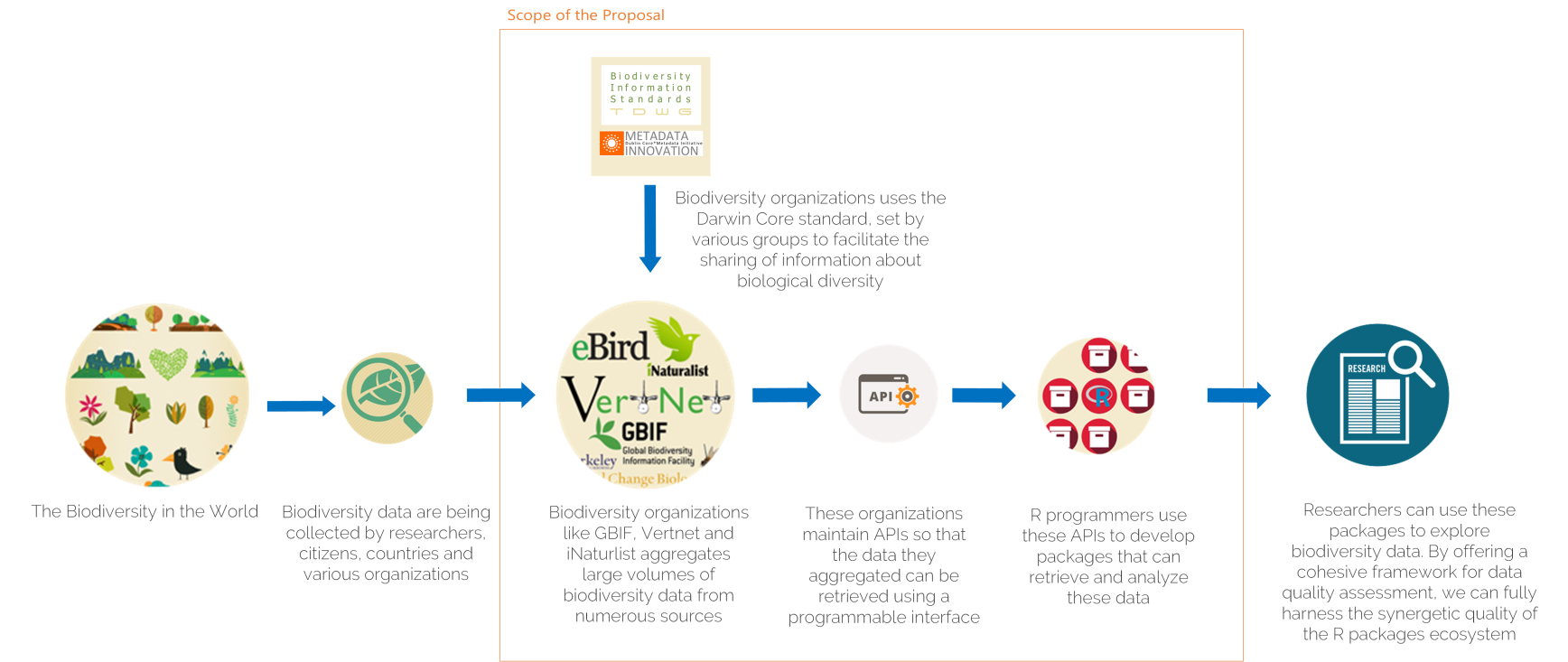
Biodiversity citizen scientists use iNaturalist to post their observations with photographs. The observations are then curated there by crowd-sourcing the identifications and other trait related aspects too. The data once converted to “research grade” is passed on to GBIF as occurrence records.
Exciting news from India in 3rd week of April 2019 is:
Another important milestone in #Biodiversity Citizen Science in #India. This week we crossed 100K verifiable records on @inaturalist this data is about ~10K species by 4500+ observers #CitSci pic.twitter.com/DCF3QxQl1i
— Vijay Barve (@vijaybarve) April 21, 2019
Being interested in biodiversity data visualizations and completeness, I was waiting for 100k records to explore the data. Here is what I did and found out.
Step 1: Download the data from iNaturalist website. Which can be done very easily by visiting the website and choosing the right options.
https://www.inaturalist.org/observations?place_id=6681
I downloaded the file as .zip and extracted the observations-xxxx.csv. [In my case it was observations-51008.csv].
Step 2: Read the data file in R
library(readr)
observations_51008 <- read_csv("input/observations-51008.csv")
Step 3: Clean up the data and set it up to be used in package bdvis.
library(bdvis) inatc <- list( Latitude="latitude", Longitude="longitude", Date_collected="observed_on", Scientific_name="scientific_name" ) inat <- format_bdvis(observations_51008,config = inatc)
Step 4: We still need to rename some more columns for ease in visualizations like rather than ‘taxon_family_name’ it will be easy to have field called ‘Family’
rename_column <- function(dat,old,new){
if(old %in% colnames(dat)){
colnames(dat)[which(names(dat) == old)] <- new
} else {
print(paste("Error: Fieldname not found...",old))
}
return(dat)
}
inat <- rename_column(inat,'taxon_kingdom_name','Kingdom')
inat <- rename_column(inat,'taxon_phylum_name','Phylum')
inat <- rename_column(inat,'taxon_class_name','Class')
inat <- rename_column(inat,'taxon_order_name','Order_')
inat <- rename_column(inat,'taxon_family_name','Family')
inat <- rename_column(inat,'taxon_genus_name','Genus')
# Remove records excess of 100k
inat <- inat[1:100000,]
Step 5: Make sure the data is loaded properly
bdsummary(inat)
will produce some like this:
Total no of records = 100000 Temporal coverage... Date range of the records from 1898-01-01 to 2019-04-19 Taxonomic coverage... No of Families : 1345 No of Genus : 5638 No of Species : 13377 Spatial coverage ... Bounding box of records 6.806092 , 68.532 - 35.0614769085 , 97.050133 Degree celles covered : 336 % degree cells covered : 39.9524375743163
The data looks good. But we have a small issue, we have some records from year 1898, which might cause trouble with some of our visualizations. So let us drop records before year 2000 for the time being.
inat = inat[which(inat$Date_collected > "2000-01-01"),]
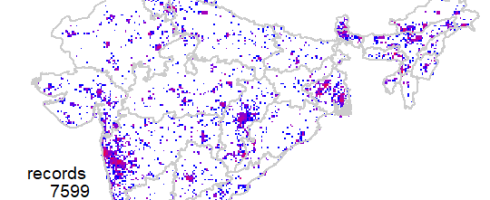
Now we are ready to explore the data. First one I always like to see is geographical coverage of the data. First let us try it at 1 degree (~100km) grid cells. Note here I have Admin2.shp file with India states map.
mapgrid(inat,ptype="records",bbox=c(60,100,5,40),
shp = "Admin2.shp")

This shows a fairly good geographical coverage of the data at this scale. We have very few degree cells with no data. How about fines scale though? Say at 0.1 degree (~10km) grid. Let us generate that.
mapgrid(inat,ptype="records",bbox=c(60,100,5,40),
shp = "Admin2.shp",
gridscale=0.1)

Now the pattern is clear, where the data is coming from.
To be continued…
References
- Barve, Vijay, and Javier Otegui. 2016. “Bdvis: Visualizing Biodiversity Data in R.” Bioinformatics. doi:http://dx.doi.org/10.1093/bioinformatics/btw333.